Computer-Aided Drug Design CADD: Types, Uses, Examples, Softwares
Table Of Content

These are important metrics for the practical sustainability of on-demand synthesis because reduced success rates or unreasonable time and cost would diminish its advantage over custom synthesis. CADD methods are mathematical tools to manipulate and quantify the properties of potential drug candidates as implemented in a number of programs. These include a range of publicly and commercially available software packages; the subset described below represents examples of fundamental tools for CADD with emphasis on those commonly used in our laboratory. Al., the blue components are a part of computer-aided drug design (CADD) and it has transformed the way targets are being discovered and validated.
Strengths and Challenges of CADD in COVID-19 Research
LBDD is generally categorized as Quantitative Structure Activity Relationship (QSAR) or pharmacophore modeling. In this review, we have briefly described about CADD and its use in the development of the therapeutic drug candidates against NDs. The successful applications, limitations and future prospects of this approach have also been discussed. Feature papers represent the most advanced research with significant potential for high impact in the field. A FeaturePaper should be a substantial original Article that involves several techniques or approaches, provides an outlook forfuture research directions and describes possible research applications. Dr. Pellecchia is a Professor of Biomedical Sciences at the School of Medicine of the University of California Riverside (UCR) and is the founding Director of the Center for Molecular and Translational Medicine at UCR.
Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders
The development of more robust generative chemical spaces can also be supported by new computational approaches in synthetic chemistry, for example, predictions of new iterative reaction sequences129 or synthetic routes and feasibility from DL-based retrosynthetic analysis130. In generative models, synthesizability predictions can be coupled with predictions of potency and other properties towards higher levels of automated chemical design131. Thus, generative adversarial networks combined with reinforcement learning (GAN-RL) were recently used to predict synthetic feasibility, novelty and biological activity of compounds, enabling the iterative cycle of in silico optimization, synthesis and testing of the ligands in vitro50,132. When applied within a set of well-established reactions and pharmacologically explored classes of targets, these approaches already yield useful hits and leads, leading to clinical candidates50,132. However, the wider potential of automated chemical design concepts and robotic synthesis in drug discovery remains to be seen.
BMC Chemistry
In addition to the common methods introduced in the first edition of this chapter, additional CADD methods developed recently in our laboratory as well as from other laboratories will be described below. The CADD Users Manual establishes the CADD standards using US Customary Units (English) and covers many of the resource files needed to complete a project within the Caltrans' right of way. This manual is subject to changes that reflect Caltrans' current development/delivery process using MicroStation and Civil3D for all projects on the State Highway System (SHS). The UC DDC was created by the University of California Biomedical Research Acceleration, Integration & Development (UC BRAID) Drugs, Devices, Diagnostics, Development (D4) group, and it has since expanded to actively include industry sponsors. Support for the consortium has been provided by a UC Office of the President (UCOP) Multi-Campus Research Proposal Initiative grant (MRPI) and its industry sponsor(s).
4 Docking-based VS
Such identified non-β-lactam-based β-lactamase inhibitors have the potential to be used in combination therapy with lactam-based antibiotics against multi-drug resistant clinical isolates. A combination of advanced computational techniques, biological science, and chemical synthesis was introduced to facilitate the discovery process, and this combinational approach enhanced the scale of discovery. Eventually, the term computer-aided drug design (CADD) was adopted for the use of computers in drug discovery [17, 18]. Advanced computational applications have been shown to be effective tools and notable successes have been achieved using these techniques. CADD is a specialized discipline, whereby different computational methods are used to simulate interactions between receptors and drugs in order to determine binding affinities [19]. However, the technique is not limited to studies of chemical interactions and binding affinity predictions, as it has many more applications ranging from the design of compounds with desired physiochemical properties to the management of digital repositories of compounds.
2. Quantitative Structure-Activity Relationships (QSARs)
We believe this review will be helpful for better understanding of CADD and its applications towards the discovery of new drug candidates against various fatal NDs. For larger system, more advanced MD techniques can be employed to enhance the sampling efficiency such as replica exchange methods. The protocols developed in our lab such as Hamiltonian replica exchange with biasing potentials (107) and replica exchange with concurrent solute scaling and Hamiltonian biasing in one dimension (108) are efficient replica exchange methods for use to enhance the MD efficiency. However, with all MD based methods the user must perform careful analysis to assure that the conformational ensemble is adequately converged for effective use in CADD. 1Conformational flexibility of molecules is a very important feature no matter if it is a small ligand or a large protein. Thus conformational sampling of a protein or ligand that produces an ensemble of biological meaningful conformations is necessary either for SBDD or for LBDD.
Libraries & institutions
Using Quantum Physics to Aid Drug Discovery - MedicalExpo e-Magazine - MedicalExpo e-Magazine
Using Quantum Physics to Aid Drug Discovery - MedicalExpo e-Magazine.
Posted: Thu, 29 Jun 2023 07:00:00 GMT [source]
The best models achieved a Spearman rank coefficient of 0.53 with a root-mean-square error of 0.95 for the predicted versus experimental pKd values in the challenge set. Such accuracy was found to be on par with the accuracy and recall of single-point experimental assays for kinase inhibition, and may be useful in screenings for the initial hits for less explored kinases and guiding lead optimization. Note, however, that the kinase family is unique as it is the largest class of more than 500 targets, all possessing similar orthosteric binding pockets and sharing high cross-selectivity. The distant second family with systematic cross-reactivity comprises about 50 aminergic GPCRs, whereas other GPCR families and other cross-reactive protein families are much smaller.
Upon target validation process, identification of hits and lead discovery phases has to be developed for a novel drug discovery process. Although, physical and biochemical parameters were used to decide the change in the structural property of the compounds to synthesize an effective lead molecule for the development of the drug. Several natural leads have become available in various databases and the literature with biological activity against its specific target, which leads to chemical modification. The most efficient and powerful process which governs entire drug discovery is structure-based drug designing (SBDD). The information about the target of small molecules, genetic information with their sequences, binding information, cytotoxicity, absorption, metabolism, excretion(ADMET) data, and other important biological information serve as the most efficient sources for accelerating the drug discovery process.
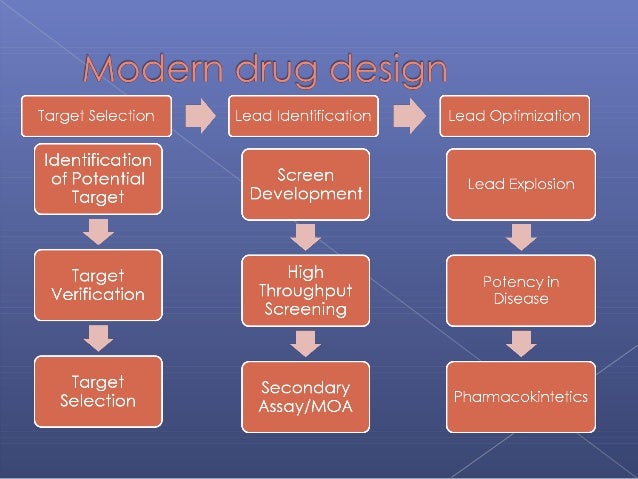
The successful application of CADD approaches for the treatment of neurodegenerative disorders is also included in this review. Following are the steps required to perform a standard MD simulation (see Note 2 for additional MD techniques). Dr. Scott R. Lokey is a Professor of Chemistry in the Department of Chemistry and Biochemistry at the University of California, Santa Cruz. His research group focuses on the relationship between molecular structure and drug-like properties, especially cell permeability. Professor Lokey is also the director of the UCSC Chemical Screening Center, a high-throughput screening facility dedicated to early stage lead discovery, and is co-founder of Circle Pharma, a biotech startup focusing on the discovery of cyclic peptide inhibitors against intracellular targets. Unfortunately, CADD suffers from cognitive dissonance (bias to seek consonance) and also sloppy research that results from the lack of proper training.
Moreover, thousands of easily synthesizable analogues assure extensive SAR-by-catalogue for the best hits, which, for example, enabled approximately 100-fold potency and selectivity improvement for the CB2 V-SYNTHES hits26. Availability of the multilayer on-demand chemical space extensions (for example, supported by MADE building blocks47) can also greatly streamline the next steps in lead optimization through ‘virtual MedChem’, thus reducing extensive custom synthesis. The limited size and diversity of screening libraries have long been a bottleneck for detection of novel potent ligands and for the whole process of drug discovery. An average ‘affordable’ high-throughput screening (HTS) campaign29 uses screening libraries of about 50,000–500,000 compounds and is expected to yield only a few true hits after secondary validation. Those hits, if any, are usually rather weak, non-selective, have suboptimal ADMET and PK properties and unknown binding mode, so their discovery entails years of painstaking trial-and-error optimization efforts to produce a lead molecule with satisfying potency and all the other requirements for preclinical development. Scaling of HTS to a few million compounds can be afforded only in big pharma, and it still does not make that much difference in terms of the quality of resulting hits.
The focused candidate ligand sets, predicted by such screening, often show useful (10–40%) hit rates in experimental testing60, yielding novel hits for many targets with potencies in the 0.1–10-μM range (for those that are published, at least). Further steps in optimization of the initial hits obtained from standard screening libraries of less than 10 million compounds, however, usually require expensive custom synthesis of analogues, which has been afforded only in a few published cases20,61. Identifying novel, potential drugs for NDs is difficult using traditional approaches of drug discovery [7]. However, during the last decade, computers have been used to aid and accelerate the process of drug discovery, and this process is now referred to as computer-aided drug design (CADD) or computer-assisted molecular design (CAMD). Neurodegenerative disorders (NDs) are diverse group of disorders characterized by escalating loss of neurons (structural and functional). The development of potential therapeutics for NDs presents an important challenge, as traditional treatments are inefficient and usually are unable to stop or retard the process of neurodegeneration.
His research laboratory focuses on the design of novel pharmacological tools and therapeutics in oncology, neurodegeneration, and other disease areas. Dr. Rogawski is Professor of Neurology and Pharmacology at the University of California, Davis School of Medicine. His research encompasses discovery of neurological therapeutics, characterization of drug mechanism, and early and later stage drug development. Dr. Rogawski is an elected fellow of the American Association for the Advancement of Science and was awarded the UC Davis Chancellor's Innovator of the Year Award for inventing the drug Zulresso™.
Although allowing comprehensive space coverage, the reaction path and success rate of generated compounds are unknown, and thus require computational prediction of their practical synthesizability. More recently, virtual on-demand chemical databases (fully enumerated) and spaces (not enumerated) allow fast parallel synthesis from available building blocks, using validated or optimized protocols, with synthetic success of more than 80% and delivery in 2–3 weeks (see the figure, part b). The virtual chemical spaces assure high chemical novelty and allow fast polynomial growth with the addition of new synthons and reaction scaffolds, including 4+ component reactions.
In its most common form, it involves modification of a known active scaffold or linking known active scaffolds, although de novo drug design (i.e., from scratch) is also possible. Though highly interrelated, identification of active scaffolds should be conceptually separated from drug design. Traditionally, the drug design process has focused on the molecular determinants of the interactions between the drug and its known or intended molecular target. Most antibiotics were designed to target proteins involved in intracellular processes, thus the outer membrane of bacteria needs to be penetrated for antibiotics to function.
The recent examples of hybrid fragment-based computational design approaches targeting SARS-CoV-2 inhibitors highlight the challenges presented by such targets and allow head-to-head comparisons to ultra-large VLS. One of the studies was aimed at the SARS-CoV-2 NSP3 conserved macrodomain enzyme (Mac1), which is a target critical for the pathogenesis and lethality of the virus. Building on crystallographic detection of the low-affinity (180 μM) fragments weakly binding Mac1 (ref. 139), merging of the fragments identified a 1-μM hit, quickly optimized by catalogue synthesis to a 0.4-μM lead140. In the same study, an ultra-scale screening of 400 million REAL database identified more than 100 new diverse chemotypes of drug-like ligands, with follow-up SAR-by-catalogue optimization yielding a 1.7-μM lead140. For the SARS-CoV-2 main protease Mpro, the COVID Moonshot initiative published results of crystallographic screening of 1,500 small fragments with 71 hits bound in different subpockets of the shallow active site, albeit none of them showing in vitro inhibition of protease even at 100 μM (ref. 141). Numerous groups crowdsourcing the follow-up computational design and screening of merged and growing fragments helped to discover several SAR series, including a non-covalent Mpro inhibitor with an enzymatic IC50 of 21 μM.



Comments
Post a Comment